
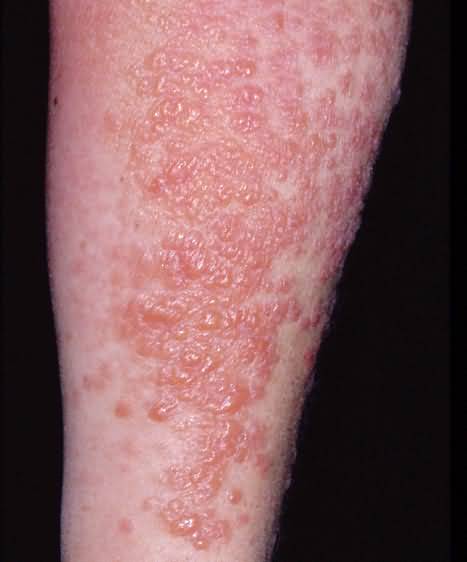












Bullous Pemphigoid: Definition
An inflammatory disease of older people; signs include widespread erythematous macules and patches, urticarial papules and plaques, and vesicles and bullae, the blisters often arising on urticarial plaques. Sometimes lesions are localized, e.g., to the legs. The cause is not known, but the mechanism is thought to be immunologic in response to antibodies to bullous pemphigoid antigens.
Bullous Pemphigoid: Adjunctive Diagnostic Tests
Direct immunofluorescence of perilesional skin shows a linear band of IgG and C3 at the epidermal basement membrane. In 70 percent of patients with bullous pemphigoid, indirect immunofluorescence reveals circulating antibasement membrane IgG.
Bullous Pemphigoid: Course
Some patients with bullous pemphigoid have urticarial papules and plaques for months before a single vesicle or bulla appears. Most patients, however, display both urticarial and blistering lesions from the outset. As a rule, the disease tends to become worse in time, i.e., the extent of distribution of lesions, the number of lesions, and the size of bullae increasing. Although the condition may wax and wane, it usually persists, if untreated, for the patient’s lifetime.
Bullous Pemphigoid: Integration: Unifying Concept
Bullous pemphigoid is a single pathologic process that manifests itself as urticarial papules and plaques and as vesicles and bullae that are subepidermal and replete with eosinophils. Although the same fundamental lesions also are present in patients with herpes gestationis, the distribution and configuration of the lesions are different. So, too, is the age of the patients; bullous pemphigoid was known formerly as “bullous disease of the aged.” The basis for bullous pemphigoid is immunologic, the disease presumably resulting from autoantibody-mediated disruption of adhesion between basal keratocytes and basement membrane, a consequence of the binding by antibodies to antigens of bullous pemphigoid that are involved in attaching basal keratocytes to the basement membrane. The antigens are bullous pemphigoid-antigen 1 (BPAG1; 230kD) which belongs to a family of genes that includes desmoplakin and bullous pemphigoid antigen 2 (BPAG2; Overview
Bullous pemphigoid is usually a self-limited inflammatory subepidermal blistering disorder of the elderly, in which circulating auto antibodies to epitopes in the basement membrane zone bind to this region and initiate a cascade of inflammatory events resulting in urticarial and bullous lesions. The therapeutic strategy is to reduce auto antibody production and to control the inflammatory reaction. Because bullous pemphigoid is not life threatening and agents that reduce auto antibody formation are inherently high risk, the initial goal of therapy is to control inflammation and reduce blister formation to a level consistent with individual patient comfort.
Initial Steps
1. Treat with oral prednisone alone at 40 to 60 mg/ day in a single morning dose. A dramatic clinical response will be seen in 70 to 80% of patients after 2 to 3 weeks. 2. Clobetasol ointment or another superpotent topical steroid is applied to the affected areas twice daily.
Subsequent Steps
Over 2 to 8 weeks, taper the prednisone dose to 30 mg/day by reduction of the daily dose by 10 mg every 2 to 3 weeks. Then, over the next 2 to 3 months, reduce prednisone to 10 to 20 mg daily. After several months, shift the patient to alternate steroid therapy, reaching 10 mg every other day. When 10 mg every other day is reached, taper dose by 1 mg every other day every 1 to 4 weeks until the lowest amount of prednisone controlling the appearance of new lesions is reached.
Alternative Steps
Alternative agents may be used in patients who do not rapidly respond to systemic steroids or in patients in whom the use of systemic steroids is contraindicated.
1. If the patient has complications or side effects from the prednisone, has persistent severe disease, or if sufficient clinical response is not achieved after 3 weeks, begin an immunosuppressive agent in combination with prednisone. In each case, continue the immunosuppressive agent for at least 2 months before abandoning it as ineffective. Once the steroid-sparing agent has begun to work, the dose of systemic steroids is gradually tapered following the protocol above. The choice of immunosuppressive agent is determined by the patient’s coexistent medical conditions, the risk of complications, and the cost. There is no evidence that one agent is superior to another. Steroid-sparing immunosuppressives include:
a. Mycophenolate mofetil 500 mg to 1.5 mg twice daily (1.0 to 3.0 g daily)
b. Azathioprine 100 to 150 mg/day
c. Methotrexate 5mg to 25 mg once weekly as a single oral dose
d. Cyclophosphamide 50 to 150 mg/day
2. Adjunctive measures that may be added to prednisone or prednisone plus immunosuppressive agents. a. Tetracycline 500 mg three or four times daily plus niacinamide 500 mg three times daily. This may be adequate alone to control mild disease or may provide additional antiinflammatory effect without causing additional immunosuppression.
b. Plasmapheresis to remove circulating pemphigoid autoantibody may be of value, but should be combined with cyclophosphamide 50 to 150 mg to inhibit antibody production.
c. Intravenous immunoglobin infusions (IVIG) may be used in refractory cases. The dose is 2g/kg given over 3 to 5 days. This is very costly and is less frequently required than in pemphigus vulgaris.
Complications and Undesired Consequences
1. Patients with bullous pemphigoid are often elderly and frail. They tolerate high-dose systemic steroid therapy poorly. Do not be reluctant to add low doses of a steroid-sparing agent to allow for reduction in the systemic steroid dose. Complications usually occur when systemic steroid dose is not reduced judiciously.
2. Potential complications of systemic steroids and immunosuppressants must be carefully sought in all patients so treated.
3. Other bullous diseases such as epidermolysis bullosa acquisita, erythema multiforme, and bullous drug eruptions may clinically resemble pemphigoid and may explain atypical therapeutic responses.
4. Immunosuppression may result in reactivation of herpes simplex or varicella zoster virus infection. These bullous infectious diseases may be generalized and simulate a flare of the pemphigoid.
180kD; Type VII collagen).
Herpes Gestationis: Definition
An inflammatory disease of the second and third trimesters of pregnancy characterized by widespread urticarial papules and plaques accompanied by vesicles and bullae. Individual lesions are indistinguishable from those of bullous pemphigoid, clinically and histopathologically. Although the cause is not known, the mechanism is thought to be immunologic in response to antibodies to an antigen peculiar to pregnancy.
Herpes Gestationis: Adjunctive Diagnostic Test
Studies of peribullous and urticarial lesions by direct immunofluorescence reveal linear deposits of C3 and IgG (in about 30 percent of patients) along the basement membrane zone.
Herpes Gestationis: Course
The disease does not begin in a first pregnancy, but in a subsequent one and then usually in each pregnancy thereafter. The urticarial and blistering lesions tend to erupt during the second trimester and then come and go until parturition, at which time they either cease altogether or sputter for a few more days before being extinguished (until the next pregnancy).